
Lessons learned from accelerating foyer with dask
Published:
Combining Foyer + Dask
More into the foray of combining modern molecular modeling tools with modern data science libraries…
Foyer uses graph algorithms to parametrize your molecular model
Given a system of molecules and atoms, how do we parametrize each atom according to our molecular model, our force field? The parameters for each atom depend on its bonded neighbors. Framing this as a graph problem (vertices are atoms and edges are bonds), subgraph isomorphisms are used to match our atom’s bonding patterns to the template bonding patterns specified by our force field’s atom-type bonding patterns
Dask helps distribute parallel workloads
Generally, most of these molecular modeling packages operate on a shared memory data structure - a list, a dictionary. To parallelize this atomtyping operation, we need to identify how we can parallelize this. For graph problems, sometimes each node (atom) needs to know every other node. We are left with a couple options
- Broadcast the entire molecular graph to all workers, divy up which atoms each worker is reponsible for atomtyping. This risks some large overhead because the entire molecular graph can span tens of thousands (or more) nodes.
- Broad only the relevant molecular graph to each worker, each worker becomes responsible for parametrizing that small subgraph. This one doesn’t involve broadcasting large graphs, but now the problem becomes identifying what the relevant graph is. I refer readers to the concept of a graph component
What to expect in this notebook
First, I’ll be breaking up the entire chemical system into smaller subgraphs. I’ll try to atom-type each subgraph serially. Then, I’ll try to distribute the workload of each subgraph using dask. I’ll try to do some timings - against different numbers of homogeneous molecules and different numbers of heterogeneous molecules. Along the way, I’ll be observing some friction points for using dask (casual user here) and for using foyer/parmed
Parallelization’s value is hard to demonstrate in this use case
Dask did not show improvements compared to canonical foyer. With the data structures we, and foyer, usually deal with, there’s some extra work in formatting them into easily-distributable data structures for parallelization. There’s always communication issues for parallel workloads. Foyer has molecule caching that accelerates atom-typing for molecules you’ve already atom-typed; this isn’t leveraged well in a distributed scenario. Foyer uses networkx, which likely already comes with its own optimizations for simplifying the workload, so evaluating a singular large graph may not be as bad as we think compared to lots of small graphs. As written, the foyer code may be best utilized serially. Future foyer implementations and refactors might better exposed elements of parallelization
Distributing the workload: split a chemical system into smaller components, parametrize each molecule, in serial
Use mbuild to create our molecule, replicate to 10 molecules, foyer to apply the OPLS-AA force field
import mbuild as mb
from mbuild.lib.recipes import Alkane
import foyer
import parmed as pmd
import networkx as nx
_ColormakerRegistry()
ff = foyer.forcefields.load_OPLSAA()
/home/ayang41/programs/foyer/foyer/forcefield.py:449: UserWarning: No force field version number found in force field XML file.
'No force field version number found in force field XML file.'
/home/ayang41/programs/foyer/foyer/forcefield.py:461: UserWarning: No force field name found in force field XML file.
'No force field name found in force field XML file.'
/home/ayang41/programs/foyer/foyer/validator.py:132: ValidationWarning: You have empty smart definition(s)
warn("You have empty smart definition(s)", ValidationWarning)
single = Alkane(n=5)
cmpd = mb.fill_box(single, n_compounds=10, box=[10,10,10])
/home/ayang41/programs/mbuild/mbuild/compound.py:2139: UserWarning: No simulation box detected for mdtraj.Trajectory <mdtraj.Trajectory with 1 frames, 3 atoms, 1 residues, without unitcells>
"mdtraj.Trajectory {}".format(traj)
/home/ayang41/programs/mbuild/mbuild/compound.py:2139: UserWarning: No simulation box detected for mdtraj.Trajectory <mdtraj.Trajectory with 1 frames, 4 atoms, 1 residues, without unitcells>
"mdtraj.Trajectory {}".format(traj)
/home/ayang41/programs/mbuild/mbuild/compound.py:2527: UserWarning: No box specified and no Compound.box detected. Using Compound.boundingbox + 0.5 nm buffer. Setting all box angles to 90 degrees.
"No box specified and no Compound.box detected. "
view = single.visualize(backend='nglview')
view
NGLWidget()
structure = cmpd.to_parmed()
Box of pentanes as parmed structures
import nglview
nglview.show_parmed(structure)
NGLWidget()
Creating the molecule graph for all moleucles in our system
graph = nx.Graph()
graph.add_nodes_from([a.idx for a in structure.atoms])
graph.add_edges_from([(b.atom1.idx, b.atom2.idx) for b in structure.bonds])
Here we can see there’s a few different graph connected components here, AKA each molecule
import matplotlib
%matplotlib inline
import matplotlib.pyplot as plt
fig, ax = plt.subplots(1,1, figsize=(8,8), dpi=100)
nx.draw_networkx(graph, node_size=100, with_labels=False, ax=ax)

Fortunately, networkx API has a connected components implementation. We have a list of sets of atom indices, where each set of atom indices refers to a connected component
individual_molecule_graphs = [*nx.connected_components(graph)]
individual_molecule_graphs[0:3]
[{0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16},
{17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33},
{34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50}]
For each individual molecule graph, we can create a parmed structure. Our entire box of pentanes was one parmed structure, but now we’re interested in creating N different parmed structures, one for each molecule. You could imagine creating another kind of object, like an mbuild compound or openmm topology, but to fit the foyer workflow, we operate on parmed structures.
all_substructures = []
for molecule_graph in individual_molecule_graphs:
individual_structure = pmd.Structure()
for idx in molecule_graph:
individual_structure.add_atom(structure.atoms[idx], structure.atoms[idx].residue.name,
structure.atoms[idx].residue.number)
for neighbor_idx in graph[idx]:
if idx < neighbor_idx:
individual_structure.bonds.append(pmd.Bond(structure.atoms[idx],
structure.atoms[neighbor_idx]))
all_substructures.append(individual_structure)
all_substructures
[<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>]
Simple iteration through each molecular subtructure, apply the force field to each
parametrized_substructures = []
for substructure in all_substructures:
output_struc = ff.apply(substructure)
parametrized_substructures.append(output_struc)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 20, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
parametrized_substructures
[<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>]
Because parmed structures override addition, we can combine structures via addition
parametrized_substructures[0] + parametrized_substructures[1]
<Structure 34 atoms; 2 residues; 32 bonds; parametrized>
Using functools, we can quickly and conveniently combine all N parametrized structures into 1 structure
from functools import reduce
parametrized_structure = reduce(lambda x,y: x+y, parametrized_substructures)
parametrized_structure
<Structure 170 atoms; 10 residues; 160 bonds; parametrized>
Rather than parametrize one, big parmed structure, we are parametrizing a bunch of small parmed structures, in serial. We’re not distributing the workload, but we are simplifying the workload – rather than match subgraphs among large, complex graphs of hundreds of nodes and edges, we are matching subgraphs among smaller, simpler graphs
Split a chemical system into smaller components, parametrize each molecule, in parallel
import dask
from dask import delayed, bag as db
Streamline our code into functions that are mostly-compatible with dask.
- The use of tuples over lists because tuples are hashable (important for dask)
- Extra functions to map atomic indices to parmed Atoms. If we’re going to create different parmed structures, we need to track parmed atoms
from typing import List, Union, Set, Dict, Tuple
def structure_to_graph(structure: pmd.Structure) -> nx.Graph:
graph = nx.Graph()
graph.add_nodes_from([a.idx for a in structure.atoms])
graph.add_edges_from([(b.atom1.idx, b.atom2.idx) for b in structure.bonds])
return graph
def separate_molecule_graphs(structure: pmd.Structure, graph: nx.Graph) -> Tuple[Tuple[int,...]]:
""" Use connected components to identify individual molecules"""
individual_molecule_graphs = (tuple(a) for a in nx.connected_components(graph))
return individual_molecule_graphs
def subselect_atoms(structure: pmd.Structure, indices: Tuple[int])-> Dict[int, pmd.Atom]:
""" Create a mapping of index to atom """
return {idx: structure.atoms[idx] for idx in indices}
def make_structure_from_graph(molecule_vertices: Tuple[int],
relevant_atoms: Dict[int, pmd.Atom],
molecule_graph: nx.Graph) -> pmd.Structure:
""" From networkx graph and individal parmed atoms, make parmed structure"""
individual_structure = pmd.Structure()
for idx in molecule_vertices:
individual_structure.add_atom(relevant_atoms[idx], relevant_atoms[idx].residue.name,
relevant_atoms[idx].residue.number)
for neighbor_idx in molecule_graph[idx]:
if idx < neighbor_idx:
individual_structure.bonds.append(pmd.Bond(relevant_atoms[idx],
relevant_atoms[neighbor_idx]))
return individual_structure
def parametrize(ff: foyer.Forcefield, structure: pmd.Structure, **kwargs) -> pmd.Structure:
return ff.apply(structure, **kwargs)
Exercising our functions in serial
We’ll get to some timings later…
%%time
single = Alkane(n=5)
cmpd = mb.fill_box(single, n_compounds=10, box=[5,5,5])
structure = cmpd.to_parmed()
big_graph = structure_to_graph(structure)
individual_molecule_graphs = separate_molecule_graphs(structure, big_graph)
individual_structures = [make_structure_from_graph(molecule_graph, subselect_atoms(structure, molecule_graph), big_graph)
for molecule_graph in individual_molecule_graphs]
parametrized_structures = [parametrize(ff, struc) for struc in individual_structures]
parametrized_structure = reduce(lambda x,y: x+y, parametrized_structures)
parametrized_structure
/home/ayang41/programs/mbuild/mbuild/compound.py:2139: UserWarning: No simulation box detected for mdtraj.Trajectory <mdtraj.Trajectory with 1 frames, 3 atoms, 1 residues, without unitcells>
"mdtraj.Trajectory {}".format(traj)
/home/ayang41/programs/mbuild/mbuild/compound.py:2139: UserWarning: No simulation box detected for mdtraj.Trajectory <mdtraj.Trajectory with 1 frames, 4 atoms, 1 residues, without unitcells>
"mdtraj.Trajectory {}".format(traj)
/home/ayang41/programs/mbuild/mbuild/compound.py:2527: UserWarning: No box specified and no Compound.box detected. Using Compound.boundingbox + 0.5 nm buffer. Setting all box angles to 90 degrees.
"No box specified and no Compound.box detected. "
CPU times: user 2.69 s, sys: 56.1 ms, total: 2.75 s
Wall time: 2.7 s
<Structure 170 atoms; 10 residues; 160 bonds; parametrized>
Here’s a first attempt at daskifying everything with delayed objects. Once we’ve created our entire system graph, we can start creating dask objects, starting with each molecule graph, and chaining the following operations:
- From each molecule graph, grab the relevant parmed Atoms
- From the molecule graph and parmed Atoms, create the (unparametrized) parmed Structure
%%time
single = Alkane(n=5)
cmpd = mb.fill_box(single, n_compounds=10, box=[5,5,5])
structure = cmpd.to_parmed()
big_graph = structure_to_graph(structure)
individual_molecule_graphs = [*separate_molecule_graphs(structure, big_graph)]
all_subselected_atoms = [delayed(subselect_atoms)(structure, molecule_graph)
for molecule_graph in individual_molecule_graphs]
raw_structures = [delayed(make_structure_from_graph)(molecule_graph, subselected_atoms, big_graph)
for molecule_graph, subselected_atoms in zip(individual_molecule_graphs, all_subselected_atoms)]
CPU times: user 149 ms, sys: 23.2 ms, total: 173 ms
Wall time: 60.4 ms
Pulse check, can we flush the task-graph and actually get our parametrized molecules?
%%time
[a.compute() for a in raw_structures]
CPU times: user 18.8 ms, sys: 889 µs, total: 19.7 ms
Wall time: 11.8 ms
[<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; NOT parametrized>]
Next step, parametrization
%%time
param_structures = [delayed(parametrize)(ff, struc) for struc in raw_structures]
CPU times: user 5.69 ms, sys: 594 µs, total: 6.28 ms
Wall time: 2.81 ms
(Another) pulse check, does the FF application work?
%%time
all_parametrized = [op.compute() for op in param_structures]
all_parametrized
CPU times: user 2.69 s, sys: 15.9 ms, total: 2.71 s
Wall time: 2.7 s
[<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>,
<Structure 17 atoms; 1 residues; 16 bonds; parametrized>]
Final step, putting the structures back together
%%time
reduce(lambda x,y: x+y, all_parametrized)
CPU times: user 52.7 ms, sys: 23 µs, total: 52.7 ms
Wall time: 50.8 ms
<Structure 170 atoms; 10 residues; 160 bonds; parametrized>
%%time
single = Alkane(n=5)
cmpd = mb.fill_box(single, n_compounds=10, box=[5,5,5])
structure = cmpd.to_parmed()
big_graph = structure_to_graph(structure)
individual_molecule_graphs = [*separate_molecule_graphs(structure, big_graph)]
all_subselected_atoms = [delayed(subselect_atoms)(structure, molecule_graph)
for molecule_graph in individual_molecule_graphs]
raw_structures = [delayed(make_structure_from_graph)(molecule_graph, subselected_atoms, big_graph)
for molecule_graph, subselected_atoms in zip(individual_molecule_graphs, all_subselected_atoms)]
param_structures = [delayed(parametrize)(ff, struc) for struc in raw_structures]
CPU times: user 65.5 ms, sys: 41.5 ms, total: 107 ms
Wall time: 51.4 ms
Last step is to combine all the parametrized structures, we can try some dask fold/reduce operations
param_structures_bag = db.from_sequence(param_structures)
param_structures_bag
dask.bag<from_sequence, npartitions=10>
Unfortuantely, some of these parmed AtomType objects are not hashable, so we cannot use dask to efficiently reduce parmed structures
from operator import add
param_structures_bag.fold(add).compute()
---------------------------------------------------------------------------
TypeError Traceback (most recent call last)
<ipython-input-37-2285eb100c4e> in <module>
1 from operator import add
2
----> 3 param_structures_bag.fold(add).compute()
...
~/miniconda3/envs/md37/lib/python3.7/site-packages/cloudpickle/cloudpickle.py in save_global(self, obj, name, pack)
828 elif obj is type(NotImplemented):
829 return self.save_reduce(type, (NotImplemented,), obj=obj)
--> 830 elif obj in _BUILTIN_TYPE_NAMES:
831 return self.save_reduce(
832 _builtin_type, (_BUILTIN_TYPE_NAMES[obj],), obj=obj)
TypeError: unhashable type: '_UnassignedAtomType'
At this point, we can use dask to parallelize most of the steps in our process, but we still need to collect all of our parametrized structures prior to summing them all up
Timing isn’t so great but we’ll see how this scales
%%time
computed_parametrized_structures = [d.compute() for d in param_structures]
final_structure = reduce(lambda x,y: x+y, computed_parametrized_structures)
final_structure
CPU times: user 2.75 s, sys: 0 ns, total: 2.75 s
Wall time: 2.74 s
<Structure 170 atoms; 10 residues; 160 bonds; parametrized>
Putting all of our parallelized code together …
%%time
# Make our molecular system
single = Alkane(n=5)
cmpd = mb.fill_box(single, n_compounds=10, box=[5,5,5])
structure = cmpd.to_parmed()
# Convert to graphs
big_graph = structure_to_graph(structure)
individual_molecule_graphs = [*separate_molecule_graphs(structure, big_graph)]
# Grab parmed atoms for each node in the graph
all_subselected_atoms = [delayed(subselect_atoms)(structure, molecule_graph)
for molecule_graph in individual_molecule_graphs]
# Generate parmed structures for each molecule
raw_structures = [delayed(make_structure_from_graph)(molecule_graph, subselected_atoms, big_graph)
for molecule_graph, subselected_atoms in zip(individual_molecule_graphs, all_subselected_atoms)]
# Parametrize with our force field
param_structures = [delayed(parametrize)(ff, struc) for struc in raw_structures]
computed_parametrized_structures = [d.compute() for d in param_structures]
final_structure = reduce(lambda x,y: x+y, computed_parametrized_structures)
CPU times: user 2.7 s, sys: 58.3 ms, total: 2.76 s
Wall time: 2.7 s
Visualizing our task graph
param_structures[0].visualize()
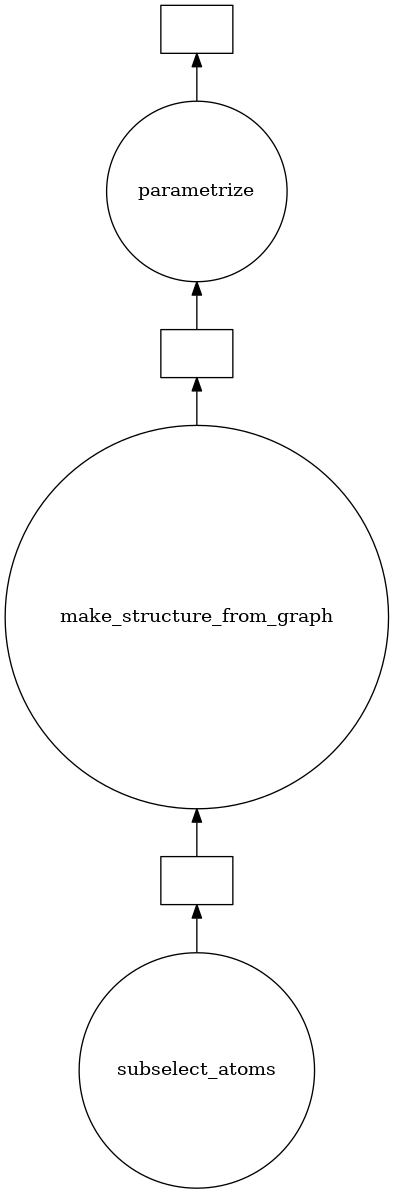
Before moving to timing comparisons, it’s important to observe the residue_map functionality for foyer. If a “residue” (molecule type) has already been parametrized within this foyer apply function stack, we don’t need to re-iterate and re-discover the atom-types; the parametrization is effectively cached. As multiple foyer apply functions get called, this caching doesn’t get leveraged.
Timing comparisons
We have 3 methods to compare:
Canonical foyer, the standard way to use foyer on a single parmed structure that represents your entire molecular system. This actualy takes most advantage of the use_residue_map functionality
Distributed foyer in serial, divide your parmed structure into smaller parmed structures, parametrize individually
Distributed foyer in parallel, divide your parmed structure into smaller parmed structures, parametrize individually.
We’ll notice the number of residues in the final, parametrized strucutres are different – this is a consequnce of how parmed.structure.__add__ and parmed.structure.__iadd__ work when you try to combine different parmed structures. What’s important is that the number of atoms and bonds are consistent
def canonical_foyer(ff, structure, **kwargs):
""" Standard way of using foyer, no parallelization"""
return ff.apply(structure, **kwargs)
def distributed_foyer_serial(ff, structure):
""" Apply foyer N times to N different molecules in serial"""
big_graph = structure_to_graph(structure)
individual_molecule_graphs = separate_molecule_graphs(structure, big_graph)
individual_structures = [make_structure_from_graph(molecule_graph, subselect_atoms(structure, molecule_graph), big_graph)
for molecule_graph in individual_molecule_graphs]
parametrized_structures = [parametrize(ff, struc) for struc in individual_structures]
parametrized_structure = reduce(lambda x,y: x+y, parametrized_structures)
return parametrized_structure
def distributed_foyer_parallel(ff, structure):
"""Apply foyer N times to N different molecules in parallel"""
big_graph = structure_to_graph(structure)
individual_molecule_graphs = [*separate_molecule_graphs(structure, big_graph)]
# Grab parmed atoms for each node in the graph
all_subselected_atoms = [delayed(subselect_atoms)(structure, molecule_graph)
for molecule_graph in individual_molecule_graphs]
# Generate parmed structures for each molecule
raw_structures = [delayed(make_structure_from_graph)(molecule_graph, subselected_atoms, big_graph)
for molecule_graph, subselected_atoms in zip(individual_molecule_graphs, all_subselected_atoms)]
# Parametrize with our force field
param_structures = [delayed(parametrize)(ff, struc) for struc in raw_structures]
computed_parametrized_structures = [d.compute() for d in param_structures]
final_structure = reduce(lambda x,y: x+y, computed_parametrized_structures)
return final_structure
Small, homogeneous system
10 pentane molecules
| Method | Time |
|---|---|
| Canonical foyer | 2.53 s |
| Distributed foyer serial | 3.15 s |
| Distributed foyer parallel | 3.22 s |
%%time
ff = foyer.forcefields.load_OPLSAA()
single = Alkane(n=5)
cmpd = mb.fill_box(single, n_compounds=10, box=[10,10,10])
structure = cmpd.to_parmed()
canonical_foyer(ff, structure)
CPU times: user 2.52 s, sys: 57.7 ms, total: 2.58 s
Wall time: 2.53 s
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 200, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
<Structure 170 atoms; 1 residues; 160 bonds; PBC (orthogonal); parametrized>
%%time
ff = foyer.forcefields.load_OPLSAA()
single = Alkane(n=5)
cmpd = mb.fill_box(single, n_compounds=10, box=[10,10,10])
structure = cmpd.to_parmed()
distributed_foyer_serial(ff, structure)
CPU times: user 3.17 s, sys: 28.5 ms, total: 3.2 s
Wall time: 3.15 s
<Structure 170 atoms; 10 residues; 160 bonds; parametrized>
%%time
ff = foyer.forcefields.load_OPLSAA()
single = Alkane(n=5)
cmpd = mb.fill_box(single, n_compounds=10, box=[10,10,10])
structure = cmpd.to_parmed()
distributed_foyer_parallel(ff, structure)
CPU times: user 3.21 s, sys: 69.5 ms, total: 3.28 s
Wall time: 3.22 s
<Structure 170 atoms; 10 residues; 160 bonds; parametrized>
Large, homogeneous system
100 pentane molecules
| Method | Time |
|---|---|
| Canonical foyer | 21.1 s |
| Distributed foyer serial | 35.1 s |
| Distributed foyer parallel | 34.7 s |
%%time
ff = foyer.forcefields.load_OPLSAA()
single = Alkane(n=5)
cmpd = mb.fill_box(single, n_compounds=100, box=[1000,1000,1000])
structure = cmpd.to_parmed()
canonical_foyer(ff, structure)
CPU times: user 20.9 s, sys: 196 ms, total: 21.1 s
Wall time: 21 s
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 2000, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
<Structure 1700 atoms; 1 residues; 1600 bonds; PBC (orthogonal); parametrized>
%%time
ff = foyer.forcefields.load_OPLSAA()
single = Alkane(n=5)
cmpd = mb.fill_box(single, n_compounds=100, box=[1000,1000,1000])
structure = cmpd.to_parmed()
distributed_foyer_serial(ff, structure)
CPU times: user 35.1 s, sys: 124 ms, total: 35.2 s
Wall time: 35.1 s
<Structure 1700 atoms; 100 residues; 1600 bonds; parametrized>
%%time
ff = foyer.forcefields.load_OPLSAA()
single = Alkane(n=5)
cmpd = mb.fill_box(single, n_compounds=100, box=[1000,1000,1000])
structure = cmpd.to_parmed()
distributed_foyer_parallel(ff, structure)
CPU times: user 34.8 s, sys: 159 ms, total: 34.9 s
Wall time: 34.7 s
<Structure 1700 atoms; 100 residues; 1600 bonds; parametrized>
Small, heterogeneous system
10 pentane, 10 decane, 10 nonadecane (C20-ane)
| Method | Time |
|---|---|
| Canonical foyer | 14 s |
| Distributed foyer serial | 16.9 s |
| Distributed foyer parallel | 16.6 s |
%%time
ff = foyer.forcefields.load_OPLSAA()
templates = [Alkane(n=5), Alkane(n=10), Alkane(n=20)]
cmpd = mb.fill_box(templates, n_compounds=[10,10,10], box=[100,100,100])
structure = cmpd.to_parmed()
canonical_foyer(ff, structure)
CPU times: user 14.2 s, sys: 130 ms, total: 14.3 s
Wall time: 14 s
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 1400, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
<Structure 1110 atoms; 1 residues; 1080 bonds; PBC (orthogonal); parametrized>
%%time
ff = foyer.forcefields.load_OPLSAA()
templates = [Alkane(n=5), Alkane(n=10), Alkane(n=20)]
cmpd = mb.fill_box(templates, n_compounds=[10,10,10], box=[100,100,100])
structure = cmpd.to_parmed()
distributed_foyer_serial(ff, structure)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 40, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 80, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
CPU times: user 17.1 s, sys: 170 ms, total: 17.3 s
Wall time: 16.9 s
<Structure 1110 atoms; 30 residues; 1080 bonds; parametrized>
%%time
ff = foyer.forcefields.load_OPLSAA()
templates = [Alkane(n=5), Alkane(n=10), Alkane(n=20)]
cmpd = mb.fill_box(templates, n_compounds=[10,10,10], box=[100,100,100])
structure = cmpd.to_parmed()
distributed_foyer_parallel(ff, structure)
CPU times: user 16.7 s, sys: 222 ms, total: 16.9 s
Wall time: 16.6 s
<Structure 1110 atoms; 30 residues; 1080 bonds; parametrized>
Large, heterogeneous system
100 pentane, 100 decane, 100 nonadecane
| Method | Time |
|---|---|
| Canonical foyer | 2 min 31 s |
| Distributed foyer serial | 4 min 20 s |
| Distributed foyer parallel | 4 min 17 s |
%%time
ff = foyer.forcefields.load_OPLSAA()
templates = [Alkane(n=5), Alkane(n=10), Alkane(n=20)]
cmpd = mb.fill_box(templates, n_compounds=[100,100,100], box=[1000,1000,1000])
structure = cmpd.to_parmed()
canonical_foyer(ff, structure)
CPU times: user 2min 30s, sys: 1.27 s, total: 2min 31s
Wall time: 2min 31s
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 14000, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
<Structure 11100 atoms; 1 residues; 10800 bonds; PBC (orthogonal); parametrized>
%%time
ff = foyer.forcefields.load_OPLSAA()
templates = [Alkane(n=5), Alkane(n=10), Alkane(n=20)]
cmpd = mb.fill_box(templates, n_compounds=[100,100,100], box=[1000,1000,1000])
structure = cmpd.to_parmed()
distributed_foyer_serial(ff, structure)
CPU times: user 4min 20s, sys: 1.05 s, total: 4min 21s
Wall time: 4min 20s
<Structure 11100 atoms; 300 residues; 10800 bonds; parametrized>
%%time
ff = foyer.forcefields.load_OPLSAA()
templates = [Alkane(n=5), Alkane(n=10), Alkane(n=20)]
cmpd = mb.fill_box(templates, n_compounds=[100,100,100], box=[1000,1000,1000])
structure = cmpd.to_parmed()
distributed_foyer_parallel(ff, structure)
CPU times: user 4min 17s, sys: 972 ms, total: 4min 18s
Wall time: 4min 17s
<Structure 11100 atoms; 300 residues; 10800 bonds; parametrized>
Random heterogeneous system
| Method | Time |
|---|---|
| Canonical foyer | 1 min 38 s |
| Distributed foyer serial | 2 min 56 s |
| Distributed foyer parallel | 3 min 1 s |
import numpy as np
random_compounds = mb.Compound(subcompounds=[Alkane(n=i) for i in np.random.randint(5, high=20, size=200)])
/home/ayang41/programs/mbuild/mbuild/compound.py:2139: UserWarning: No simulation box detected for mdtraj.Trajectory <mdtraj.Trajectory with 1 frames, 3 atoms, 1 residues, without unitcells>
"mdtraj.Trajectory {}".format(traj)
/home/ayang41/programs/mbuild/mbuild/compound.py:2139: UserWarning: No simulation box detected for mdtraj.Trajectory <mdtraj.Trajectory with 1 frames, 4 atoms, 1 residues, without unitcells>
"mdtraj.Trajectory {}".format(traj)
%%time
ff = foyer.forcefields.load_OPLSAA()
structure = random_compounds.to_parmed()
canonical_foyer(ff, structure, use_residue_map=False)
CPU times: user 1min 37s, sys: 158 ms, total: 1min 37s
Wall time: 1min 37s
<Structure 7663 atoms; 1 residues; 7463 bonds; PBC (orthogonal); parametrized>
%%time
ff = foyer.forcefields.load_OPLSAA()
structure = random_compounds.to_parmed()
distributed_foyer_serial(ff, structure)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 28, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 44, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 60, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 24, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 56, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 52, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 64, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 68, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 36, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 32, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 76, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 72, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 48, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
CPU times: user 2min 56s, sys: 839 ms, total: 2min 57s
Wall time: 2min 56s
<Structure 7663 atoms; 200 residues; 7463 bonds; parametrized>
%%time
ff = foyer.forcefields.load_OPLSAA()
structure = random_compounds.to_parmed()
distributed_foyer_parallel(ff, structure)
CPU times: user 3min 1s, sys: 551 ms, total: 3min 2s
Wall time: 3min 1s
<Structure 7663 atoms; 200 residues; 7463 bonds; parametrized>
Making individual structures
Parallelization is fantastically slowing down our operations. I have a hunch this might be due to the extra steps involved in splitting up the molecular graphs.
When molecular modelers make these systems, we already know which collection of atoms and bonds forms a molecule, so we can use that to circumvent any use of connected components. In this iteration, we’ve added a shortcut where we already know the individual structures.
Canonical foyer is still faster. For a parallel library comparison, I tried using multiprocessing but got infinite recursion errors, so multiprocessing was not as easy to use as dask for this particular application
random_compounds = mb.Compound(subcompounds=[Alkane(n=i) for i in np.random.randint(5, high=20, size=200)])
%%time
individual_structures = [cmpd.to_parmed() for cmpd in random_compounds.children]
param_structures = [delayed(parametrize)(ff, struc) for struc in individual_structures]
computed_parametrized_structures = [d.compute() for d in param_structures]
final_structure = reduce(lambda x,y: x+y, computed_parametrized_structures)
final_structure
/home/ayang41/programs/mbuild/mbuild/compound.py:2527: UserWarning: No box specified and no Compound.box detected. Using Compound.boundingbox + 0.5 nm buffer. Setting all box angles to 90 degrees.
"No box specified and no Compound.box detected. "
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 76, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 36, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 24, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 72, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 68, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 60, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 32, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 64, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 28, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 56, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 52, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 40, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 48, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 44, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
CPU times: user 3min 19s, sys: 1.04 s, total: 3min 20s
Wall time: 3min 19s
<Structure 7735 atoms; 200 residues; 7535 bonds; PBC (orthogonal); parametrized>
param_structures[0].visualize(rankdir='LR')
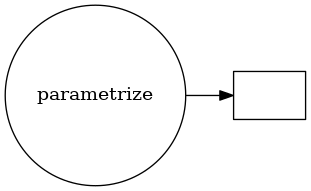
%%time
one_structure = random_compounds.to_parmed()
canonical_foyer(ff, one_structure)
/home/ayang41/programs/mbuild/mbuild/compound.py:2527: UserWarning: No box specified and no Compound.box detected. Using Compound.boundingbox + 0.5 nm buffer. Setting all box angles to 90 degrees.
"No box specified and no Compound.box detected. "
CPU times: user 1min 50s, sys: 1.05 s, total: 1min 51s
Wall time: 1min 51s
/home/ayang41/programs/foyer/foyer/forcefield.py:267: UserWarning: Parameters have not been assigned to all impropers. Total system impropers: 9780, Parameterized impropers: 0. Note that if your system contains torsions of Ryckaert-Bellemans functional form, all of these torsions are processed as propers
warnings.warn(msg)
<Structure 7735 atoms; 1 residues; 7535 bonds; PBC (orthogonal); parametrized>
Lessons and Takeaways
This was a little disheartening, any attempt to distribute foyer atom-typing or combine with dask did NOT accelerate anything. This can probably be explained in a variety of ways:
We had to convert our structure to a graph, run a connected components algorithm (which has its own scaling issues), create separate parmed structures, then re-join/add the individual structures together. Each of those steps is bound to slow things down. Data communication also plays a role here – communicating the molecular graphs and the entire structure to each dask worker will add some slowness to our pipeline. Doing everything in one foyer function allows the use of caching, which we lose when executing the function lots of different times. Even simplifying the pipeline didn’t show much improvement for the dask implementation
There probably is room for the foyer API to be more accommodating for dask and other parallel computations, but it might require a refactoring effort to properly expose the functions-to-parallelize and utilize data structures/approaches more amenable to parallelization. Breaking up a large chemical system into smaller substructures didn’t seem to help.
In all honesty since most molecular systems usually have less than a dozen different molecular species, just replicated into thousands of molecules, the best bet is to parametrize each molecular species once, then propagate the parameters appropriately, all in the canonical foyer style without any parallelization. The current foyer implementation already has implicit acceleration with caching and networkx may already have some graph optimizations for subgraph isomorphisms, mitigating any need for us to explicitly decompose one big graph into lots of small connected components
Notebooks can be found in this repo